
By Ares Cabó Carrera and Carolina Lezama (ERNI Spain)
Transforming a life science device or research-use-only (RUO) device into a medical one is a process full of opportunities and complexities. In one project, we supported a company in converting a lab diagnostics device for clinical use. This article highlights the journey in a regulated setting and how expertise drives success.
The challenge
While the core technology appeared to be the same, patient safety, legal liability, traceability and other documentation changed almost everything. The regulatory requirements, expectations and scope of controlled processes expanded vastly – and with this, the expectations on the product developers changed, shifting from speed or flexibility of development to full accountability and validated traceability.
In this instance, the challenges became evident and were multifaceted – fragmented requirements; the ever-changing needs of stakeholders; deadlines; ISO, FDA and EU MDR compliance; and the awareness of the need to prevent costly rework and compliance failure.
The process: A journey to regulation
Our approach to these challenges was requirements engineering (RE). RE provided a traceable and structured foundation from the start so we were able to keep technical portions of the development aligned with regulatory needs, manage change appropriately and effectively, and ensure that no critical detail was overlooked or forgotten. RE served to be the lever moving us from scientific discovery to clinical compliance.
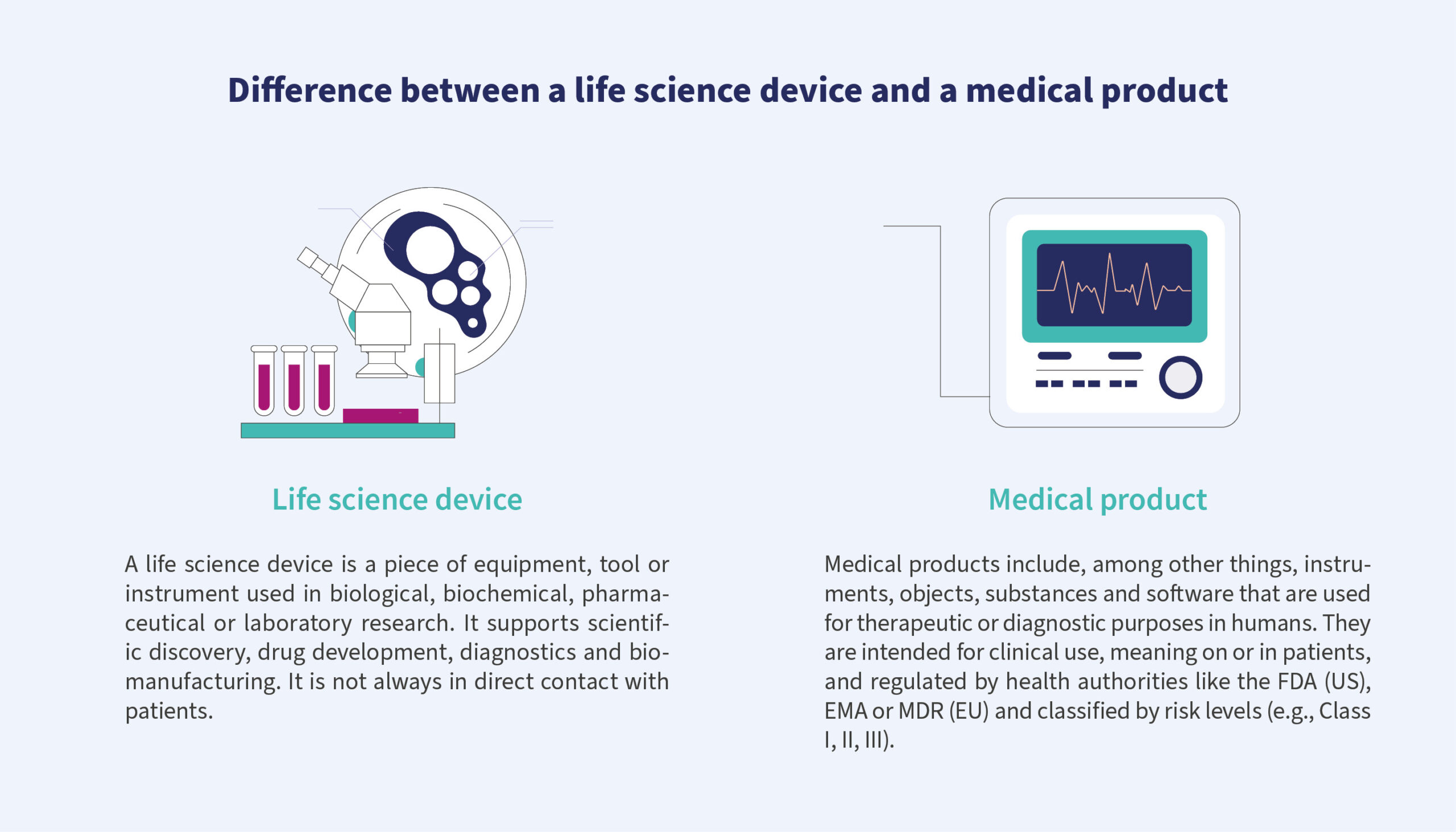
Transforming software from a life science research tool into a regulated diagnostic medical device is not a linear upgrade; it’s a fundamental shift in mindset, methodology and responsibility. This transformation journey took our teams from the relatively flexible realm of scientific software into the rigour of regulated diagnostics, where every function must be justified, every risk mitigated and every line of code traceable.
In regard to research-use-only products, teams often optimise for speed, functionality and exploration.
Documentation is less heavy, testing is pragmatic and risk management is rather informal. In contrast, a medical device – subject to FDA and IVDR requirements – demands process maturity, systematic traceability and verified quality at every stage.
That meant rethinking how we worked, from how we documented and validated user needs to how we verified software behaviour under real-world clinical conditions. Risk management was no longer a background activity; it became embedded in every backlog refinement and decision checkpoint.
Reimagining agile in a regulated world
Agile principles remained at the core of our approach, but we needed to adapt them to fit a compliance-driven environment. Instead of fast iteration at the expense of traceability, we designed a hybrid agile model that preserved adaptability while ensuring full traceability, validation and compliance.
- Creating incremental release candidates instead of one monolithic launch. The modularity in the architecture had the purpose of optimising the code and enhancing future maintenance.
- Defining minimum viable products (MVPs) that were not only functionally usable but also regulatory-compliant. This way, we were able to deliver viable results to the customer at an early stage.
- Accepting that definition of done went far beyond ‘working software’. Each user story had to be linked to validated requirements, test cases, acceptance criteria and documented verification outcomes. If it wasn’t testable and documented, it wasn’t done.
With retrospective, in a big project like this with a hundred team members distributed over multiple sites across Europe, we would choose a different agile framework – most likely SAFe – due to reasons like too many diverse teams or not all teams following the same iteration process.
Risk at the core
We integrated risk management directly into backlog refinement. Stories were sliced not just by business value or complexity, but by risk exposure and regulatory criticality. For example, data integrity-related features – such as audit logs and result traceability – were front-loaded in the roadmap due to their impact on patient safety and compliance. This helped the team prioritise under pressure, especially when the scope had to be negotiated due to tight timelines. The MVP was not the bare minimum – it was the minimum certifiable product.
Product ownership redefined
In this setting, product owners (POs) were more than feature gatekeepers. They became translators between stakeholders, compliance teams and developers, ensuring that evolving customer needs were interpreted correctly and delivered in line with regulatory expectations. POs and requirements engineers worked side-by-side to bridge clinical needs with development constraints, constantly verifying that each deliverable could withstand regulatory scrutiny – before it reached the end of a sprint, let alone the market.
Documentation: A living organism
Throughout this project, we embraced documentation not as overhead, but as a living asset – a dynamic, evolving body of knowledge that enabled safe decision making, ensured regulatory compliance and unlocked scalability. Each release Candidate was treated as a self-contained milestone. Rather than front-loading all documentation or postponing it until a final release, we built it up incrementally and consistently. Each user story tied into a broader user journey. In this way, documentation became a tool for alignment and quality, not just compliance. It allowed every stakeholder, from developers to regulatory reviewers, to understand what was built, why it mattered and how it was verified.
Solution
Together with our customer, we delivered not only a compliant and validated device but one that is modular, scalable and ready for the global market. We are happy the product is now on the market – feedback that we have received has been positive, not only from internal stakeholders but also from end users in clinical environments who now benefit from the device. Among the key lessons we learned, we would say that early alignment on product requirements is critical; agile practices must be tailored, not transplanted, into regulated environments; and above all, requirements engineering and documentation must be treated as strategic enablers – not administrative burdens.
To learn more about how to build secure digital products download also our eBook below.
